La segnalazione spontanea in Italia nel 2014 e la nuova legge sulla farmacovigilanza
Come ogni anno ecco i dati sulle segnalazioni dell’anno precedente che mostrano un ulteriore passo avanti nel momento in cui esce finalmente il nuovo decreto sulla farmacovigilanza
All’inizio di maggio 2015 il ministro della Salute Beatrice Lorenzin ha firmato il decreto di recepimento della normativa europea (direttiva 2010/84/UE) in tema di farmacovigilanza, successivamente pubblicato in Gazzetta Ufficiale (decreto ministeriale 30.04.2015, GU n. 143 del 23/06/2015). La normativa è entrata in vigore a luglio 2012 e il recepimento arriva quindi in estremo ritardo, esponendo l’Italia al rischio di pesanti sanzioni. Su Focus Farmacovigilanza abbiamo già commentato ai tempi le novità della nuova normativa (Focus, luglio 2012, pagina 2) che, nelle more del recepimento, era in pratica già entrata in vigore, soprattutto per le attività di farmacovigilanza richieste alle aziende farmaceutiche. Mancava però un chiaro riferimento legislativo, che arriva con quasi tre anni di ritardo.
I dati della segnalazione nel 2014
Prima di commentare il recepimento analizziamo brevemente la situazione della segnalazione spontanea in Italia nel 2014. Come l’anno scorso i dati in dettaglio saranno pubblicati da AIFA all’interno del Rapporto Osmed disponibile dal prossimo settembre, ma anticipiamo i dati più rilevanti sulla nostra rivista. Escludendo le oltre 4.000 segnalazioni di letteratura, nel 2014 sono state inserite nella Rete nazionale di farmacovigilanza 51.181 segnalazioni, con un incremento complessivo del 25% rispetto al 2013.
Il numero di segnalazioni per milione di abitanti varia dalle 1.652 della Lombardia alle 128 dell’Abruzzo (vedi tabella). La media nazionale è di 858 e conferma l’Italia tra i primissimi Paesi in Europa. Viene confermato la forte tendenza in crescita delle segnalazioni, che sono raddoppiate negli ultimi 3 anni. Alla quantità si associa anche la qualità, intesa come completezza delle informazioni contenute nelle segnalazioni. E’ stato recentemente pubblicato dal Centro dell’OMS UMC che l’Italia, tra i Paesi con almeno 1.000 segnalazioni/anno, è il Paese al mondo con il più alto grado di documentazione delle segnalazioni.1
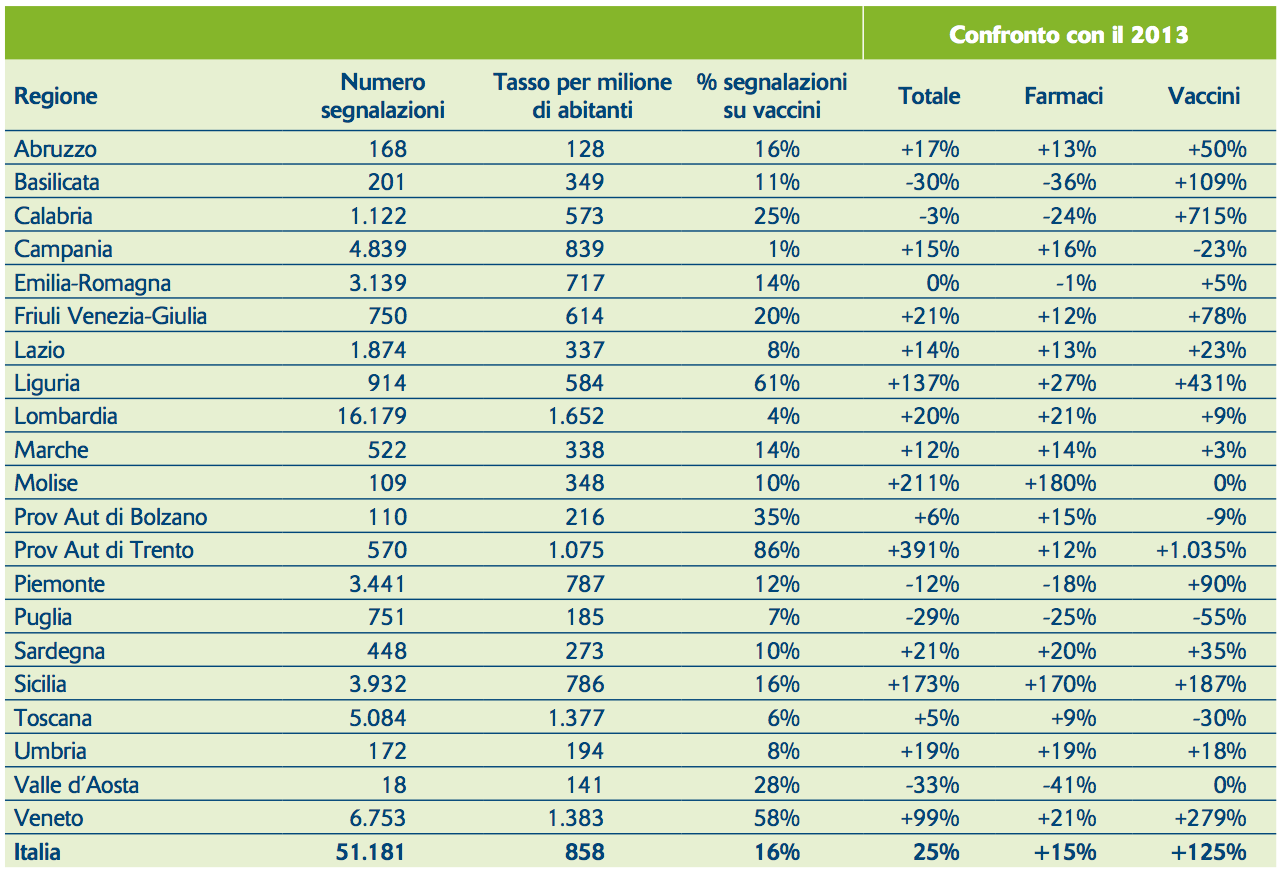
Il raddoppio delle segnalazioni da vaccino è principalmente legato a un progetto di sorveglianza degli eventi avversi da vaccino tetravalente morbillo-parotiterosolia e varicella (MPRV) che si sta svolgendo in Veneto. I risultati di questo progetto non sono ancora disponibili. Quasi la metà delle segnalazioni nel 2014 in Italia proviene da medici ospedalieri (46%). Consistente è il numero di segnalazioni da parte dei farmacisti (18%) mentre rimane bassa l’adesione al sistema da parte dei medici di medicina generale (7%) e molto scarsa (0,7%) è l’adesione al sistema da parte dei cittadini.
Come detto la situazione è influenzata dai progetti di farmacovigilanza attiva effettuati in Lombardia e in altre regioni italiane. Molti di questi interventi sono stati fatti in ambito ospedaliero e questo spiega l’alto numero di segnalazioni da questa fonte. Anche le segnalazioni dei farmacisti sono principalmente dovute a farmacisti che in ambito ospedaliero raccoglievano le segnalazioni principalmente nei reparti di Pronto soccorso. Le segnalazioni da farmacisti del territorio sono poche.
Complessivamente la percentuale di segnalazioni gravi in Italia è del 32%. Questo varia molto da regione a regione (per esempio 42% in Lombardia e 18% in Veneto) principalmente in relazione ai diversi progetti di farmacovigilanza attiva.
Quasi metà della segnalazioni (49%) riporta la risoluzione completa dell’evento avverso. Sono però presenti 381 casi che hanno avuto esito fatale, valore in linea con quanto osservato negli anni precedenti. I casi a esito fatale sono legati principalmente a farmaci anticoagulanti (warfarin o nuovi anticoagulanti) e antiaggreganti, con reazioni di sanguinamento avvenute in pazienti anziani.
Sono però presenti 58 casi fatali collegati alla vaccinazione antinfluenzale, 33 dei quali legati alla specialità Fluad®. Tutte le segnalazioni al riguardo sono state inviate negli ultimi due mesi dell’anno. Il caso delle reazioni fatali alla vaccinazione antinfluenzale ha avuto ampio risalto sui media alla fine dell’anno scorso. Tutto è partito dalla segnalazione di tre decessi avvenuti tra il 7 e il 18 novembre scorso attribuiti al vaccino antinfluenzale adiuvato Fluad®. Il vaccino viene dato soprattutto a pazienti anziani in genere con polipatologie e in politrattamento e che presentano quindi una elevata mortalità. Gli eventi segnalati erano soprattutto cardiovascolari, e il nesso di causalità con la vaccinazione era molto debole. Nonostante questo l’AIFA decise di comunicare la sospensione dei lotti collegati a queste segnalazioni, facendo fare all’Istituto Superiore di Sanità (ISS) i test necessari alla valutazione della sicurezza del vaccino. Il clamore legato a questa decisione ha causato l’invio di molte altre segnalazioni a esito fatale nei giorni successivi al primo ritiro, coinvolgendo altri lotti di vaccino. I test eseguiti dall’ISS e la valutazione dei casi ricevuti condotta in collaborazione con l’EMA ha alla fine escluso un coinvolgimento del vaccino nei casi segnalati e a fine anno l’AIFA ha tolto il divieto di utilizzo per i lotti coinvolti. Purtroppo questa vicenda ha avuto un impatto fortemente negativo: milioni di persone hanno rinunciato alla vaccinazione con conseguente forte incremento dei casi di influenza (e aumento della mortalità). Il caso del Fluad® ha avuto ripercussioni anche sul recepimento della normativa europea.
Il nuovo decreto sulla farmacovigilanza
Il nuovo decreto sulla farmacovigilanza, come detto in precedenza, ha recepito i cambiamenti imposti dalla nuova normativa europea. Alcune delle novità introdotte con la normativa erano già presenti in Italia, per esempio i pazienti possono segnalare direttamente le reazioni avverse dal 1991; altre, come la definizione di reazione avversa, sono state inserite nel testo del decreto.
Una novità importante riguarda le attività e il ruolo dei Centri regionali di farmacovigilanza. Commentando in questi anni la tendenza positiva del tasso di segnalazione abbiamo più volte sottolineato come i Centri regionali siano stati determinanti nel raggiungere questi risultati. I Centri hanno potuto usufruire dei fondi provenienti dall’AIFA collegati alla legge 449 del dicembre 1997 e distribuiti in seguito a specifici Accordi Stato-Regioni, ma hanno dovuto operare nel sistema in accordo alle linee guida emanate dall’AIFA che ha già iniziato attività di audit su questa operatività. Tutte queste funzioni si sono basate sul testo degli Accordi Stato-Regioni, ma non avevano alla base un riferimento chiaro nel decreto legge sulla farmacovigilanza che si limitava a riportare che “le Regioni si possono avvalere di Centri regionali”.
Il nuovo decreto stabilisce un riferimento chiaro sia all’operatività sia al finanziamento dei Centri che potranno quindi, sulla base di questa normativa, cercare di strutturare meglio il proprio personale, in larga parte ancora basato su contratti a tempo determinato. L’unico commento del ministro della salute sul nuovo decreto riportato dai giornali e dai siti web si riferisce alla variazione dei tempi richiesti agli operatori sanitari (ma non ai pazienti) per fare la segnalazione. Il provvedimento prevede che gli operatori sanitari debbano segnalare:
- entro due giorni le sospette reazioni avverse da medicinali;
- non oltre le 36 ore da quando ne sono venuti a conoscenza le sospette reazioni avverse da medicinali di origine biologica (per esempio i vaccini).
Questa norma, fortemente voluta dallo stesso ministro a seguito della vicenda del Fluad®, è molto criticabile per i seguenti motivi:
- una tempistica così stringente non corrisponde a quanto stabilito dalla normativa europea attualmente in vigore. Il vincolo temporale era presente nel decreto legge sulla farmacovigilanza del 1997 (tre giorni per le reazioni gravi e sei giorni per le altre) ed era stato poi tolto nella formulazione attuale al primo recepimento della normativa europea nel 2003. Si fa quindi un passo indietro;
- il tempo che intercorre tra l’insorgenza della reazione e la compilazione della segnalazione può variare in funzione del tipo di evento. In caso di eventi gravi, con pazienti che hanno diverse patologie e assumono molti farmaci la valutazione del ruolo causale del farmaco può richiedere l’esecuzione di esami clinici o di laboratorio che necessitano di tempi adeguati;
- la differenza tra 36 ore e 2 giorni non è valutabile sulla base delle informazioni riportate sulla scheda, dove compaiono la data di insorgenza della reazione e la data di compilazione;
- la tempistica proposta per la segnalazione è molto distante da quanto avviene nella pratica. Se analizziamo le segnalazioni pervenute nel 2014, solo nel 18% dei casi è stata fatta entro due giorni dall’insorgenza della reazione.
Non sappiamo come reagiranno i segnalatori all’inserimento di questo limite. Pur non essendo previste delle sanzioni, si teme possa disincentivare la segnalazione. Inoltre non impedirebbe, in presenza di un nuovo allarme, l’invio di segnalazioni effettuate in ritardo. Probabilmente rimarrà una delle tante norme ampiamente disattese presenti in Italia (al riguardo vedi anche nel nostro sito all’indirizzo: http://www.farmacovigilanza.eu/ content/se-36-o-48-ore-vi-sembranpoche%E2%80%A6).
Nonostante questa ultima variazione, che può essere considerata una ulteriore conseguenza negativa del caso Fluad®, il recepimento presenta comunque molti punti positivi, soprattutto nell’organizzazione del sistema della segnalazione spontanea in Italia ed è un buon punto di partenza per migliorare ulteriormente il sistema di farmacovigilanza in Italia.
L’anno scorso nel commentare i dati sottolineavamo la necessità di intervenire per ridurre le segnalazioni su carta. L’AIFA, in collaborazione con il Centro regionale di farmacovigilanza del Veneto, si sta decisamente muovendo in questa direzione.
In Veneto la segnalazione via web è partita da febbraio 2014 (www.vigifarmaco.it) e in meno di un anno le segnalazioni inviate online hanno superato il 20% del totale.
L’esperienza si sta allargando ad altre regioni per una breve fase pilota prima di partire ufficialmente su tutto il territorio nazionale.
- Drug Saf 2014;37:65-77. CDI