Sicurezza dei farmaci: di chi la responsabilità
Quando un farmaco viene commercializzato si sa molto della sua qualità, della farmacologia e della sua efficacia in un numero attentamente selezionato di pazienti arruolati nei trial clinici. Questi dati, comunque, possono fornire un quadro incompleto e anche fuorviante dell’efficacia reale del farmaco una volta utilizzato nella popolazione generale. Ancor meno si sa, al momento dell’approvazione alla commercializzazione, riguardo alla sua sicurezza nella popolazione generale, viste le dimensioni limitate dei trial clinici e il fatto che i soggetti inclusi sono stati selezionati. In altre parole il suo bilancio globale rischi-benefici è incerto.
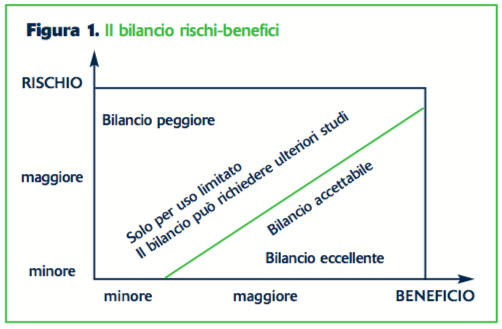
E’ importante considerare il bilancio rischi-benefici di un farmaco piuttosto che limitarsi alla sua sola sicurezza (figura 1).1 Molti farmaci con un profilo di sicurezza apparentemente terribile (per esempio i farmaci anticancro) sono usati con successo da mani esperte per la loro efficacia e hanno quindi un rapporto rischi-benefici favorevole.
Diversi protagonisti hanno responsabilità nel garantire la sicurezza del farmaco dopo la sua commercializzazione.
1. Professionisti sanitari
La responsabilità di chi prescrive un farmaco include la registrazione di qualunque danno esso possa provocare, ma anche di allargare le conoscenze del suo rapporto rischiobeneficio. La documentazione del rischio usa soprattutto tecniche di sorveglianza passiva per generare segnali. La segnalazione di reazioni avverse viene fatta alle autorità regolatorie che possono intraprendere numerose azioni. Anzitutto devono fornire un feedback ai medici che prescrivono. In secondo luogo esse possono diffondere avvertimenti (per esempio il Black Box warning negli Stati Uniti). In terzo luogo possono limitare la prescrizione a certi gruppi di pazienti oppure consentirla solo a medici specializzati. Infine le autorità possono revocare l’autorizzazione alla commercializzazione del farmaco.
C’è poi l’arricchimento delle conoscenze di farmacovigilanza. Per allargare le conoscenze sulla sicurezza di un farmaco si usano studi osservazionali o, più raramente, trial clinici usando tecniche di sorveglianza attiva. Si può definire la sorveglianza attiva come “un approccio sistematico di popolazione alla sorveglianza sulla sicurezza di un farmaco che mira ad accertare esaustivamente il numero di eventi avversi attraverso un processo continuo predefinito”.
Per tali studi i dati clinici vengono raccolti dalle cartelle cliniche dei pazienti (per esempio database di cartelle elettroniche o, in Gran Bretagna, lo UK Clinical Practice Research Datalink), da registri di pazienti (per esempio il registro della British Rheumatological Society di tutti i pazienti in terapia con farmaci antinfiammatori anti TNF) o, raramente, da trial clinici nei quali gli esiti valutati siano relativi alla sicurezza del farmaco.
2. Autorità regolatorie
La regolamentazione dei farmaci mira ad assicurare la sicurezza delle medicine e nel corso degli anni alcuni eventi chiave hanno portato a nuove norme.2
L’adulterazione di un elisir a base di una sulfonamide con etilenglicole (usato al posto del glicerolo) portò gli Stati Uniti nel 1937 a prendere provvedimenti per assicurare la qualità delle medicine. Il disastro della talidomide negli anni sessanta ha condotto alla richiesta di avere prove sulla sicurezza e l’efficacia di un farmaco prima della commercializzazione. Più recentemente, le conseguenze della somministrazione dell’anticorpo monoclonale TG1412 a volontari in un trial clinico di fase 1 ha portato a nuove norme per gli studi fatti per la prima volta nell’uomo con nuove molecole.
Tutte le autorità regolatorie aggiornano periodicamente i loro requisiti di sicurezza. In Europa nel 2012 sono state introdotte nuove norme di farmacovigilanza che hanno sottolineato l’importanza dei piani di risk management per tutti i nuovi prodotti approvati, hanno migliorato le basi legali per studi di sicurezza e di efficacia dopo la commercializzazione e hanno cercato di migliorare la trasparenza e l’accesso alle informazioni sulla sicurezza dei farmaci, che erano state finora proprie solo delle autorità regolatorie ma che sono state ora estese ad altre parti interessate.
Negli Stati Uniti il Food and Drugs Administration Amendments Act del 2007 ha definito i requisiti post commercializzazione e gli obblighi per i detentori delle autorizzazioni al commercio. Questi requisiti possono comprendere informazioni ottenute attraverso la sorveglianza di eventi avversi, studi osservazionali e trial clinici. La legge definisce anche le strategie di valutazione e riduzione del rischio (REMS) che possono essere applicate alle nuove medicine o a medicine per le quali sono stati osservati nuovi rischi per la sicurezza. La legge propone anche un nuovo sistema di farmacovigilanza attiva battezzato Sentinella. Sono stati stabiliti programmi mini-sentinella come preliminari rispetto ai più grandi progetti sentinella. Questi programmi sono studi pilota che usano dati elettronici (per esempio quelli degli Electronic Health Records, EHRs) e dati di segnalazione per valutare la sicurezza dei farmaci in commercio e dei dispositivi medici. Al settembre 2013 il Mini-Sentinel Database conteneva informazioni su 360 milioni di anni-persona e 4 miliardi di prescrizioni farmaceutiche. E’ stato proposto che i programmi sentinella forniscano informazioni sulla sicurezza successiva alla commercializzazione e sulla efficacia dei farmaci in commercio e che siano anche usati per la valutazione dei segnali. Questo promette di essere un database di ricerca per una farmacoepidemiologia di alta qualità.
3. Industria
L’industria ha la responsabilità per legge di segnalare alle autorità regolatorie tutti gli effetti avversi da farmaco che gli operatori sanitari o i cittadini segnalino. E’ interessante notare che nel Regno Unito la maggior parte dei pazienti tende a segnalare tali eventi ai medici, mentre negli Stati Uniti queste segnalazioni vengono più spesso fatte direttamente all’industria. A parte ciò, i Periodic Safety Update Reports (PSURs) in Europa e i Periodic Adverse Experience Reports (PADERs) negli Stati Uniti sono revisioni periodiche sui dati di sicurezza dei farmaci. Essi ora sono confluiti nei Periodic Benefit Risk Evaluation Reports (PBRERs) che devono essere completati dall’industria e inviati alle autorità regolatorie. I PBRERs analizzano i rischi nel contesto dei benefici e della gravità delle condizioni trattate, riflettendo la convinzione che i dati di sicurezza dovrebbero essere rimpiazzati da una valutazione rischi-benefici di cui si è discusso prima.
4. Paziente
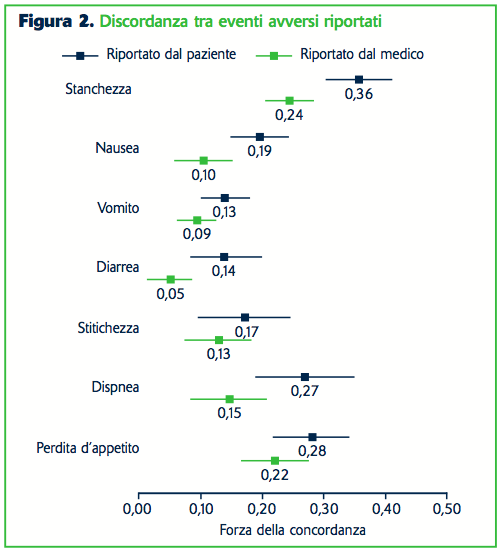
Il paziente è il più importante protagonista per quanto riguarda la sicurezza dei farmaci, ma sfortunatamente la sua voce spesso è inascoltata. I pazienti vorrebbero naturalmente sapere quali sintomi potrebbero essere dovuti al farmaco che è stato loro prescritto. Leggendo la sezione “Effetti collaterali” sul foglietto illustrativo del prodotto i pazienti trovano una valanga di informazioni. Ma ciò che non possono sapere è che queste informazioni vengono per lo più da trial clinici e sono basate quasi del tutto sulle impressioni e sull’interpretazione dei sintomi dei pazienti da parte dei medici piuttosto che da segnalazioni di prima mano di esperienze del paziente con il farmaco. Ci sono ormai prove corpose che questa valutazione da parte del medico possa essere sbagliata e che i medici sistematicamente sottostimano la gravità dei sintomi riferiti dal paziente. I report da parte dei pazienti frequentemente segnalano effetti collaterali che i medici non hanno rilevato. La figura 2 illustra la non concordanza tra rilevazione di eventi avversi da parte di medici e pazienti in un gruppo di 467 pazienti con varie forme di cancro trattati allo Sloan Kettering Institute di New York.
Non ci sono requisiti regolatori per i quali dovrebbero essere usati i report dei medici piuttosto che quelli dei pazienti nei dati dei trial clinici; tutto quello che chiedono le autorità regolatorie è che gli sponsor forniscano dati di sicurezza durante lo sviluppo e l’approvazione di un farmaco. Paradossalmente le autorità regolatorie incoraggiano che vengano riportate misure di esito riferite dal paziente durante lo sviluppo di un farmaco per sostenere le avvertenze riportate nel foglietto illustrativo. E’ come se ci fosse una mancanza di strumenti per raccogliere dati dal paziente durante lo sviluppo di un farmaco e dopo la commercializzazione. Questo è particolarmente rilevante per i disturbi soggettivi. Uno strumento importante che viene sempre più usato va sotto il capitolo dei social media. Questo può essere definito come “l’interazione sociale tra persone nella quale esse creano, condividono o scambiano informazioni e idee in comunità virtuali e network”. I social media dipendono dallo sviluppo di tecnologie mobili e dall’uso di Internet; differiscono dai media tradizionali o industriali sotto molti punti di vista, inclusa la qualità, la portata, l’usabilità e la permanenza.
Ci sono molti tipi di progetti collaborativi nei social media (per esempio Wikipedia), blog e microblog (per esempio Twitter) o siti di reti sociali (per esempio Facebook) e comunità di contenuti (per esempio You Tube), ma la differenza tra questi diventa via via sempre meno definita. Un punto importante per la presente discussione è che le agenzie regolatorie stanno col tempo realizzando che i social media possono essere metodi di comunicazione tra le comunità alle quali in precedenza non avevano accesso. E’ stato stimato che circa il 10% delle interazioni in varie forme di social media riguardano la salute e tra queste una componente importante sono i farmaci.
Per questo molte agenzie regolatorie (EMA, FDA e Agenzie nazionali) stanno ora investendo pesantemente su questa forma di comunicazione e stanno usandola non solo per diffondere importanti messaggi di allerta sulla sicurezza, ma anche per ascoltare le discussioni sugli argomenti correnti e invitare a fornire feedback. Un’area importante che sta emergendo dagli scambi nell’ambito dei social media è la reale efficacia clinica delle medicine. Nel corso degli anni Agenzie regolatorie, politici e stampa hanno posto una grande enfasi sulla sicurezza dei farmaci piuttosto che sulla loro efficacia nella popolazione generale dopo l’autorizzazione alla commercializzazione. Una volta che l’efficacia è stata stabilita nei trial clinici si pensa che sia scontata nell’uso in comunità. Le conversazioni nei social media stanno mettendo in discussione questo atteggiamento.
Conclusioni
La valutazione sulla sicurezza o ancor meglio sul rapporto rischi-benefici, è una responsabilità condivisa. I prescrittori, l’industria e i pazienti devono tutti interagire con le autorità regolatorie usando gli strumenti più rilevanti oggi disponibili. Occorre porre un’attenzione crescente alla voce dei pazienti così come occorre ancora realizzare a pieno l’impatto delle tecniche dei social media. Poiché i metodi di autorizzazione alla commercializzazione si evolvono, portando le medicine a essere disponibili precocemente sul mercato nel loro ciclo vitale, il ruolo di queste forme di valutazione sarà sempre più importante.
- Clin Pharm Therap 2009;85:241-6. CDI
- Clin Pharm Therap 2012;92:486-93. CDI
Emeritus Professor of Clinical Pharmacology,
University of Liverpool, former chairman of MHRA